



Ги запознаваме ретките болести: ХЕРЕДИТАРНА СПАСТИЧНА ПАРАПЛЕГИЈА
ГИ ЗАПОЗНАВАМЕ РЕТКИТЕ БОЛЕСТИ 2026
 Во месецот со најмалку дена во годината, во месецот кој и оваа година е редок по своите ретки 28 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по ОСМИ пат ќе се обидеме да се сретнеме со храбрите приказни и поблиску да Ве запознаеме со сите оние кои живеат со предизвиците и проблемите кои се невидливи. Ајде да започнеме нова страница во разбирањето и емпатијата, бидејќи сите заедно ја имаме силата на заедништвото, силата на надежта, силата на информирањето и силата да ги обоиме сенките носејќи светлина и видливост…
Во месецот со најмалку дена во годината, во месецот кој и оваа година е редок по своите ретки 28 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по ОСМИ пат ќе се обидеме да се сретнеме со храбрите приказни и поблиску да Ве запознаеме со сите оние кои живеат со предизвиците и проблемите кои се невидливи. Ајде да започнеме нова страница во разбирањето и емпатијата, бидејќи сите заедно ја имаме силата на заедништвото, силата на надежта, силата на информирањето и силата да ги обоиме сенките носејќи светлина и видливост…

ХЕРЕДИТАРНА СПАСТИЧНА ПАРАПЛЕГИЈА/ СТРАМПЕЛ–ЛОРЕИНОВА БОЛЕСТ/ HEREDITARY SPASTIC PARAPLEGIA/ STRÜMPELL–LORRAIN DISEASE
MKБ-10 G11.4
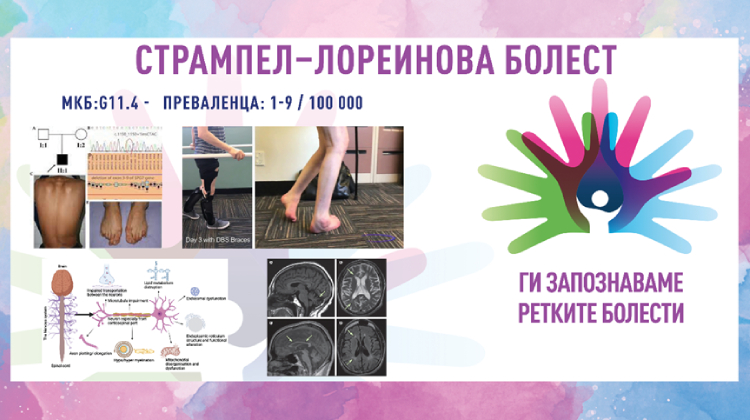 ПРЕВАЛЕНЦА – забележана кај 1–9/100 000 население; во Европа се движи од 1/11 000 до 1/77 000 население.
ПРЕВАЛЕНЦА – забележана кај 1–9/100 000 население; во Европа се движи од 1/11 000 до 1/77 000 население.
ВОВЕД
Хередитарна спастична параплегија (ХСП), позната и како Стрампел–Лореинова болест, претставува високо хетерогена група на наследни болести чија главна карактеристика е прогресивно нарушување на одењето. Болеста се манифестира со прогресивна вкочанетост (спастичност) и контракција на долните екстремитети. Симптомите се резултат на дисфункција на долгите аксони во ᾿рбетниот мозок. Зафатените клетки се примарните моторни неврони, па затоа се работи за болест на горниот моторен неврон. Болеста е предизвикана од дефекти во транспортот на протеините, структурните протеини, протеините за одржување на клетките, липидите и другите супстанции низ клетката. Долгите нервни влакна (аксоните) се засегнати бидејќи долгите растојанија ги прават нервните клетки особено чувствителни на дефекти во споменатите механизми. Започнува во детството или раната адолесценција и прогресира бавно во текот на животот. Болеста првпат била опишана во 1880 година од страна на германскиот невролог Адолф Штрумпел, додека поопширно е опишана во 1888 година од страна на францускиот лекар Морис Лорен. Терминот „хередитарна спастична параплегија“ бил измислен од страна на Анита Хардинг во 1983 година.
ЕТИОЛОГИЈА
Стрампел–Лореиновата болест се должи на дисфункција на горните моторни неврони на кортикоспиналниот тракт, пренесувајќи се на наследен начин, најчесто автосомно доминантно, но може да биде и автосомно рецесивно. Тоа значи дека болеста може да се наследи од еден или двајца родители, зависно од типот на мутацијата. Најчесто е поврзана со мутации во гените SPAST (2p22.3), ATL1 (14q22.1), REEP1 (2p11.2) и KIF5A (12q.13.3) за автосомно доминантни HSPs и SPG7 (16q24.3), SPG11 (15q21). 1), и CYP7B1 (8q12.3) за автосомно рецесивен, кои кодираат протеини важни за одржување на нормалната функција на моторните неврони во спиналниот мозок. Досега се идентификувани повеќе од 80 гени одговорни за настанокот на болеста. Кодираните протеини се вклучени во многу процеси, вклучувајќи аксонален транспорт, миелинизација, трговија со ендомембрани, функции на митохондриите, комплексен липиден и нуклеотиден метаболизам. Овие мутации доведуваат до дегенерација на кортикоспиналните неврони и оштетување на спиналните нервни патишта, со што е нарушена трансмисијата потребна за контрола на движењата на мускулите на долните екстремитети. Сепак, значителен број пациенти се без генетска дијагноза по спроведено систематско тестирање.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Хередитарна спастична параплегија се манифестира со прогресивно влошување на мускулната сила во нозете, како и со зголемена спастичност. Главните симптоми вклучуваат:
- спастичност – уште во раниот стадиум на болеста, пациентите почнуваат да забележуваат зголемена мускулна напнатост и тешкотии во движечките активности, како што се одењето или подигнувањето на нозете;
- спастичност на долните екстремитети – постепено слабеење на мускулите на нозете, што доведува до ограничена мобилност или до потреба од помагала за одење;
- ефекти во координацијата и рамнотежата – рамнотежата на телото е нарушена, а пациентите можат да имаат тешкотии во одењето, што води до чести падови;
- болка во мускулите и зглобовите – може да се јави хронична болка поради мускулната напнатост;
- прогресивно влошување – болеста обично има бавен прогресивен тек, но може да варира во зависност од конкретната генетска варијанта и возраст на појавата.
Клинички, херидитарните спастични параплегии можат да се поделат на чиста и комплицирана (сложена) форма.
Чистата форма на херидитарната спастична параплегија се карактеризира со бавно прогресивна спастичност и слабост на екстремитетите, најчесто поврзана со уринарни нарушувања и длабоки сензорни аномалии (намалена чувствителност на вибрација на долните екстремитети).
Сложената форма на херидитарната спастична параплегија се одликува со присуство или отсуство на дополнителни невролошки карактеристики. Невролошките карактеристики можат да вклучуваат нарушувања на церебелумот (атаксија, нистагмус, тремор), демиелинизирачка периферна невропатија (сензорни и/или моторни нарушувања), когнитивни нарушувања, сензорни дефекти, епилепсија, миопатски карактеристики, екстрапирамидални карактеристики (Паркинсонизам, хореа, дистонија), психијатриски нарушувања итн., кои можат да бидат индикативни за одреден генетски поттип. Отсуството на невролошките манифестации вклучува офталмолошки абнормалности (катаракта, ретинитис пигментоза, макуларна дегенерација) и ортопедски абнормалности (сколиоза, дислокација на зглобот и различни деформитети на стапалото).
Дијагнозата се поставува врз основа на клиничките симптоми, невролошкиот преглед, прогресивниот тек на болеста, мерењето на биомаркери, MРИ на мозокот и ᾿рбетот (со цел да се исклучат другите чести невролошки состојби), фамилијарната историја, молекуларното генетско тестирање и исклучувањето на другите диференцијални дијагнози, како што се: мултиплекс склероза, спинални васкуларни абнормалности, дефицит на витамин Б12, инфекција со HTLV-I, примарна латерална склероза, диплегична церебрална парализа, метаболички генетски заболувања, нарушување на акумулацијата на метали во мозокот. Крајната потврда на дијагнозата може да се обезбеди само со спроведување на генетски тестови насочени кон познати генетски мутации. Особено генетско тестирање е потребно во случаите каде што претходно била идентификувана мутација во семејството.
ПОСЛЕДИЦИ
Поголемиот дел од лицата со хередитарна спастична параплегија имаат нормален животен век. Симптомите на Стрампел–Лореиновата болест прогресивно се влошуваат со текот на времето, што може да доведе до ограничена мобилност, зголемен ризик од повреди, потреба од помагала. Постојат и компликации, односно хронична болка, деформации на екстремитетите и психосоцијални последици. Прогнозата зависи од фенотипот, генотипот, како и од самата експресија на гените. Во некои случаи, засегнатите лица се сериозно оневозможени, додека во други, засегнатите се способни да ги извршуваат повеќето секојдневни активности без потреба од прилагодувања.
ТРЕТМАН
Засега не постои специфичен третман кој би го спречил, забавил или би го променил текот на болеста. Терапијата е симптоматска и третманите се насочени кон ублажување на симптомите и подобрување на квалитетот на живот преку: физиотерапија и рехабилитација, медикаментозен третман (лекови против спастичност, аналгетици за ублажување на болката), хируршка интервенција (во некои случаи, за корекција на деформации), психолошка поддршка и слично. Од лекови, се препорачуваат:
- баклофен – мускулен релаксант за релаксација на мускулите и намалување на тонусот, кој може да се администрира орално или интратекално;
- дијазепам и клоназепам – за намалување на интензитетот на спазмите;
- оксибутинин хлорид и толтеродин тартарат – неволен мускулен релаксант и спазмолитички агенс, кој се користи за намалување на спастичноста на мочниот меур кај пациенти со проблеми со контролата на мочниот меур;
- кросистем – за намалување на мускулната прекумерна активност;
- антидепресиви – за пациенти кои доживуваат клиничка депресија.
Особено важна е физикалната терапија која помага во обновувањето и одржувањето на способноста за движење, исто така помага за намалување на мускулниот тонус, за одржување или подобрување на опсегот на движење и мобилност, за да се зголеми силата и координацијата, за да се спречат компликации, како што се замрзнати зглобови, контрактури или рани.
за проектот:
Здружението на пациенти Ретко е да си редок од Охрид и овој февруари, во рамките на осмата сезона од проектот ГИ ЗАПОЗНАВАМЕ РЕТКИТЕ БОЛЕСТИ 2026, Ве запознаваат со ретките болести од кои заболува населението во Р.С. Македонија, со надеж дека преку овие нови 28 ретки дијагнози сите заедно ќе успееме да го кренеме гласот за проблемите со кои се соочуваат лицата со ретки болести, но и нивните семејства.
Кампањата е од информативно – едукативен карактер и на неа работеа над 70 еминентни професори и доктори од Клиничкиот центар Мајка Тереза од Скопје, доктори од здравствените установи од нашиот охридско – струшки регион – Охридската и Струшката болница, ЈЗУ Специјална болница за ортопедија и трауматологија “Св. Еразмо” – Охрид, ЈЗУ Специјализирана болница за превенција, лекување и рехабилитација на кардиоваскуларни заболувања “Свети Стефан” – Охрид, како и доктори од многу приватни ординации од општините во Р.С. Македонија. Она што навистина загрижува е бројноста и различноста на ретките болести и информациите за навистина ултра ретки дијагнози како што се Fahr’s Syndrome, Jalili syndrome, Marcus-Gunn syndrome, MoyaMoya disease, Shukla-Vernon syndrome…
Сепак надежта останува и понатаму да не води и искрено се надеваме дека ова истражување ќе ни помогне да изработиме база на податоци која ќе ни послужи за планирање на следните активности во интерес на лицата со ретки болести, на семејствата меѓусебно поврзување и размена на искуства. Она што не охрабрува и не мотивира да одиме напред е позитивниот ефект од кампањите кои ги водевме во изминатите години, свесни сме дека помогнавме и охрабривме многу пациенти и семејства да зборуваат јавно за својата болест, да соберат храброст да не исконтактираат, а секако добивме многу пофалби и од стручната медицинска фела која е сè поактивно вклучена во кампањите, а и ние упатуваме јавна благодарност затоа што без нивната помош и стручните совети немаше да успееме.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29-ти февруари, РЕДОК датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има РЕДОК број на денови. Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008-ма година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година. Р. С. Македонија за прв пат се вклучи во одбележување на овој редок ден во 2012-та година. Кампањата за Денот на Ретки Болести е за пошироката јавност, за релевантните институции и секој е добредојден да се приклучи: пациенти и семејства, организации на пациенти, медицински професионалци, истражувачи, фармацевтска индустрија, јавни здравствени институции – колку повеќе не има, толку подобро. Денот на ретките болести е можност за учесниците да бидат дел од глобалниот повик кон креаторите на политики, истражувачите, компаниите и здравствените професионалци повеќе и поефикасно да ги вклучат пациентите во истражувањата за ретки болести.
ПРИДРУЖИТЕ НИ СЕ!
StrugaOnline







