



Ги запознаваме ретките болести: ШВАРЦ–ЏАМПЕЛОВ СИНДРОМ
ГИ ЗАПОЗНАВАМЕ РЕТКИТЕ БОЛЕСТИ 2026
 Во месецот со најмалку дена во годината, во месецот кој и оваа година е редок по своите ретки 28 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по ОСМИ пат ќе се обидеме да се сретнеме со храбрите приказни и поблиску да Ве запознаеме со сите оние кои живеат со предизвиците и проблемите кои се невидливи. Ајде да започнеме нова страница во разбирањето и емпатијата, бидејќи сите заедно ја имаме силата на заедништвото, силата на надежта, силата на информирањето и силата да ги обоиме сенките носејќи светлина и видливост…
Во месецот со најмалку дена во годината, во месецот кој и оваа година е редок по своите ретки 28 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по ОСМИ пат ќе се обидеме да се сретнеме со храбрите приказни и поблиску да Ве запознаеме со сите оние кои живеат со предизвиците и проблемите кои се невидливи. Ајде да започнеме нова страница во разбирањето и емпатијата, бидејќи сите заедно ја имаме силата на заедништвото, силата на надежта, силата на информирањето и силата да ги обоиме сенките носејќи светлина и видливост…

ШВАРЦ–ЏАМПЕЛОВ СИНДРОМ/ХОНДРОДИСТРОФИЧНА МИОТОНИЈА/ SCHWARTZ–JAMPEL SYNDROME (SJS)/CHONDRODYSTROPHIC MYOTONIA
MKБ-10 Q78.8, G71.1
 ПРЕВАЛЕНЦА – забележана кај помалку од 1/100 000 население (во литературата до денес се опишани 130 случаи).
ПРЕВАЛЕНЦА – забележана кај помалку од 1/100 000 население (во литературата до денес се опишани 130 случаи).
ВОВЕД
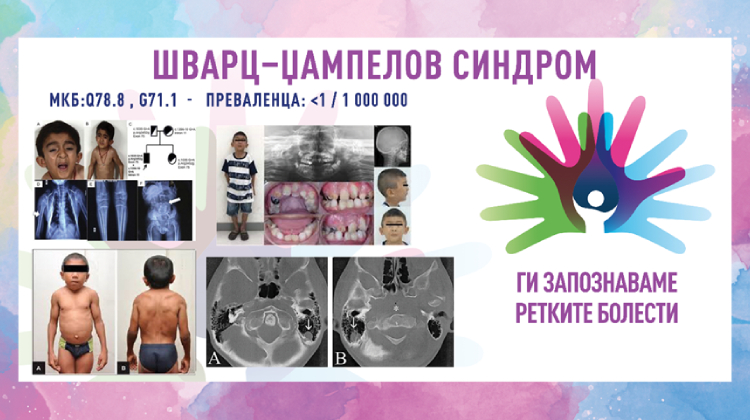
Шварц–Џампеловиот синдром (SJS) или хондродистрофична миотонија е ретка генетска болест предизвикана од мутација на генот на перлекан (HSPG2), што предизвикува остеохондродисплазија поврзана со миотонија. Тоа е генетски условен синдром кој ги вклучува мускулите и нервниот систем, и кој води до значителни физички и невролошки нарушувања, особено во поглед на мускулниот тонус и контролата на движењата. Типичните карактеристики на болеста вклучуваат ниска висина, абнормалности на скелетот, постојани мускулни контракции и хипертрофија на мускулатурата. Синдромот првпат е опишан во 1962 година од американскиот офталмолог Оскар Шварц и американскиот невроофталмолог Роберт Стивен Џампел. Поради својата реткост, оваа болест не е широко позната, и иако пациентите имаат речиси нормален животен век, сепак синдромот има сериозно влијание врз квалитетот на животот на пациентите.
ЕТИОЛОГИЈА
Шварц–Џампеловиот синдром е предизвикан од мутации во генот HSPG2, кој го кодира протеинот перлекан позициониран во мускулите и ᾿рскавицата. Перлеканот е важен за функцијата на мускулите и нервите бидејќи ги регулира јонските канали кои се вклучени во преносот на нервните импулси. Кај синдромот постои сомневање дека абнормалната функција на перлекан доведува до недостаток на ацетилхолинестераза, ензим вклучен во разградувањето на невротрансмитерот ацетилхолин, кој ја поттикнува мускулната контракција. Ако ацетилхолинот не се разложи, може да доведе до продолжена мускулна контракција/зацврстување на мускулите (миотонија). Шварц–Џампеловиот синдром се наследува автосомно рецесивно, што значи дека се потребни две копии од мутираниот ген (една од секој родител) за да се развие болеста, иако некои пријавени случаи укажуваат на автосомно доминантна шема на наследување. Родителите на детето со автосомно рецесивна состојба обично не боледуваат од таа состојба. Генетскиот локус е мапиран на 1p36.1 Некои деца од опишаните случаи во литературата имале благи скелетни промени (SJS-1), додека други имале примарна коскена дисплазија придружена со грчеви, односно миотонија (SJS-2). Првата група покажала поврзаност со хромозом 1p36.1-p34, додека втората група не покажува таква поврзаност.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Симптомите на Шварц–Џампеловиот синдром вклучуваат широк спектар на физички и невролошки проблеми. Главните симптоми се:
- миотонија – тешкотија при опуштање на мускулите по нивната контракција; се манифестира со грчење на мускулите по изведување на движења, изразито се влошува при ниска надворешна температура или при стрес;
- мускулни спазми, покачен мускулен тонус – предизвикува затегнатост на мускулите и ограничувања во движењето, како и појава на хипертрофија на мускулите на телото;
- прогресивна слабост – најчесто се јавува неподносливост на физичка активност и слабост на мускулите, особено на екстремитетите;
- очи – овој синдром може да биде поврзан со катаракта, блефароспазам (спазам на капакот) и други визуелни нарушувања;
- кардиоваскуларни проблеми – иако не се толку чести, сепак можат да се појават, како на пример, абнормалности во работата на срцето;
- коскени и зглобни деформитети – краток врат, конвексен граден кош, абнормална кривина на пршлените, криво стапало и/или рамни стапала, деформитет на колкот што вклучува зголемен агол меѓу вратот и телото на бутната коска, неправилност на капителарните епифизи на бутната коска, хондродисплазија, остеопороза.
SJS-1 – соодветствува на оригиналниот опис на Шварц и Џампел. Коскените деформитети не се истакнати при раѓање, миотонијата на лицето е главната карактеристика која создава специфична тријада што вклучува стеснување на очните прорези (блефарофимоза/блефароспазам), собирање на устата и брчкање на брадата. Стимулацијата, како на пример, лесен удар или дување врз очните капаци, предизвикува блефароспазам. Континуираната миотонија на екстремитетите предизвикува вкочанетост при одење и нетолеранција на физичка активност. Иако, моторниот развој за време на првата година е бавен, сепак интелигенцијата е нормална.
SJS-2 – се карактеризира со изразени коскени деформитети при раѓање, кои сугерираат на Моркиов синдром (остеохондродистрофија). Неонаталната смртност кај оваа форма е висока.
Шварц–Џампеловиот синдром се дијагностицира врз основа на карактеристичните црти на лицето, скелетните карактеристики и миотонијата. Тестовите на крвта можат да покажат покачена серумска креатин киназа или алдолаза. Рендгенските зраци, мускулната биопсија или електромиографијата (ЕМГ) можат да бидат од помош за засегнатите лица. Електромиографијата (ЕМГ) е специфична и клучна при поставувањето на дијагнозата и покажува континуирана активност на моторните единици. Првенствено, оваа активност се интерпретирала како миотонија. Миотонија може да биде присутна, но континуираната активност на моторните единици е одговорна за симптомите на лицето и екстремитетите. Серумската концентрација на креатин киназа (CK) може да биде благо зголемена. Хистолошкиот изглед на мускулот обично е нормален, но може да покаже варијации во големината на мускулните влакна и зголемен број на централни јадра. Генетското тестирање за генот HSPG2 може да ја потврди дијагнозата.
ПОСЛЕДИЦИ
Доколку болеста се стабилизира по адолесценцијата, тогаш истата не влијае на нормалниот животен век. Сепак, Шварц–Џампеловиот синдром има сериозни последици за пациентите и тие обично вклучуваат хронични и прогресивни невролошки и мускулни проблеми:
- хронична мускулна слабост – пациентите често имаат тешкотии во движењето, што може да доведе до потешкотии во извршувањето на секојдневни активности; мускулната слабост може да биде прогресивна, што значи дека со текот на времето ќе се зголемува потребата од помош при извршување на обичните задачи;
- проблеми со видот – честа појава се катаракта, блефароспазам (неволен спазам на капакот) и други визуелни нарушувања, и тие можат значително да го нарушат видот и квалитетот на животот на пациентите;
- проблеми со функцијата на срцето – кај некои пациенти можат да се појават кардиоваскуларни компликации, како што се абнормалности во срцевиот ритам или срцевите дефекти;
- психолошки ефекти – постојаната мускулна болка, ограниченото движење и хроничниот замор можат да доведат до депресија, анксиозност и други психолошки нарушувања, со што дополнително се влошува состојбата на пациентот;
- влијание на животен век – самата болест има тенденција да се стабилизира во адолесцентниот период и не влијае врз животниот век, но во ретки случаи, тешките последици од Шварц–Џампеловиот синдром можат да доведат до скратен животен век, особено ако сериозните компликации не се третираат навремено.
ТРЕТМАН
Третманот на Шварц–Џампеловиот синдром во голема мера е насочен кон ублажување на симптомите и подобрување на квалитетот на животот на пациентите. Пошироко, третманот вклучува комбинација на лекови, физиотерапија и хируршка интервенција во зависност од сериозноста на симптомите. Главните пристапи за третман вклучуваат:
- лекови за миотонија – лекови, како што се дијазепам или други антиконвулзивни лекови, често се користат за намалување на миотонијата (тешкотијата при опуштање на мускулите по нивната контракција); овие лекови помагаат во олеснување на грчевите и ја намалуваат мускулната затегнатост;
- физиотерапија и рехабилитација – за да се спречи влошување на мускулната слабост и да се подобри мобилноста, пациентите обично следат програма за физиотерапија; физиотерапевтите користат различни техники за вежби и мобилизации на мускулите и зглобовите, што помага во одржување на нивната функција;
- третман на очни нарушувања – може да се јави присуство на катаракта и на други проблеми со видот; катарактата може да се отстрани со хируршки третман, што може значително да го подобри видот;
- психолошка поддршка – поради психолошките ефекти кои произлегуваат од хроничната болка и ограничената мобилност, пациентите имаат потреба од терапија за поддршка при нападите на депресија и анксиозност, како што се когнитивно-бихејвиоралната терапија и советувањето;
- генетско советување – поради генетската природа на синдромот, генетското советување е од помош за семејствата кои можат да бидат засегнати од болеста; тоа им помага на родителите да ги разберат шансите за пренесување на болеста на следните генерации и да донесат паметни одлуки;
- контрола на други компликации – за да се спречат или контролираат компликациите, како што се проблеми со срцето или дијабетес, пациентите можат да бидат лекувани со специјализирани лекови и редовни контроли.
Шварц–Џампеловиот синдром е сериозна, прогресивна болест која има големо влијание на физичкото и психолошкото здравје на пациентите. Иако не постои лек, раното откривање, соодветната терапија и мултидисциплинарниот пристап за третман можат да им помогнат на пациентите да го контролираат напредувањето на болеста и да го подобрат квалитетот на животот. За пациентите со Шварц–Џампеловиот синдром, интегрираниот третман и поддршката од страна на медицинскиот тим се клучни за постигнување добри резултати.
за проектот:
Здружението на пациенти Ретко е да си редок од Охрид и овој февруари, во рамките на осмата сезона од проектот ГИ ЗАПОЗНАВАМЕ РЕТКИТЕ БОЛЕСТИ 2026, Ве запознаваат со ретките болести од кои заболува населението во Р.С. Македонија, со надеж дека преку овие нови 28 ретки дијагнози сите заедно ќе успееме да го кренеме гласот за проблемите со кои се соочуваат лицата со ретки болести, но и нивните семејства.
Кампањата е од информативно – едукативен карактер и на неа работеа над 70 еминентни професори и доктори од Клиничкиот центар Мајка Тереза од Скопје, доктори од здравствените установи од нашиот охридско – струшки регион – Охридската и Струшката болница, ЈЗУ Специјална болница за ортопедија и трауматологија “Св. Еразмо” – Охрид, ЈЗУ Специјализирана болница за превенција, лекување и рехабилитација на кардиоваскуларни заболувања “Свети Стефан” – Охрид, како и доктори од многу приватни ординации од општините во Р.С. Македонија. Она што навистина загрижува е бројноста и различноста на ретките болести и информациите за навистина ултра ретки дијагнози како што се Fahr’s Syndrome, Jalili syndrome, Marcus-Gunn syndrome, MoyaMoya disease, Shukla-Vernon syndrome…
Сепак надежта останува и понатаму да не води и искрено се надеваме дека ова истражување ќе ни помогне да изработиме база на податоци која ќе ни послужи за планирање на следните активности во интерес на лицата со ретки болести, на семејствата меѓусебно поврзување и размена на искуства. Она што не охрабрува и не мотивира да одиме напред е позитивниот ефект од кампањите кои ги водевме во изминатите години, свесни сме дека помогнавме и охрабривме многу пациенти и семејства да зборуваат јавно за својата болест, да соберат храброст да не исконтактираат, а секако добивме многу пофалби и од стручната медицинска фела која е сè поактивно вклучена во кампањите, а и ние упатуваме јавна благодарност затоа што без нивната помош и стручните совети немаше да успееме.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29-ти февруари, РЕДОК датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има РЕДОК број на денови. Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008-ма година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година. Р. С. Македонија за прв пат се вклучи во одбележување на овој редок ден во 2012-та година. Кампањата за Денот на Ретки Болести е за пошироката јавност, за релевантните институции и секој е добредојден да се приклучи: пациенти и семејства, организации на пациенти, медицински професионалци, истражувачи, фармацевтска индустрија, јавни здравствени институции – колку повеќе не има, толку подобро. Денот на ретките болести е можност за учесниците да бидат дел од глобалниот повик кон креаторите на политики, истражувачите, компаниите и здравствените професионалци повеќе и поефикасно да ги вклучат пациентите во истражувањата за ретки болести.
ПРИДРУЖИТЕ НИ СЕ!
StrugaOnline







